






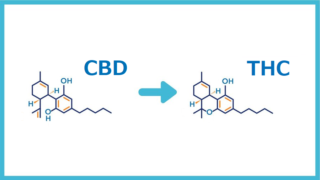











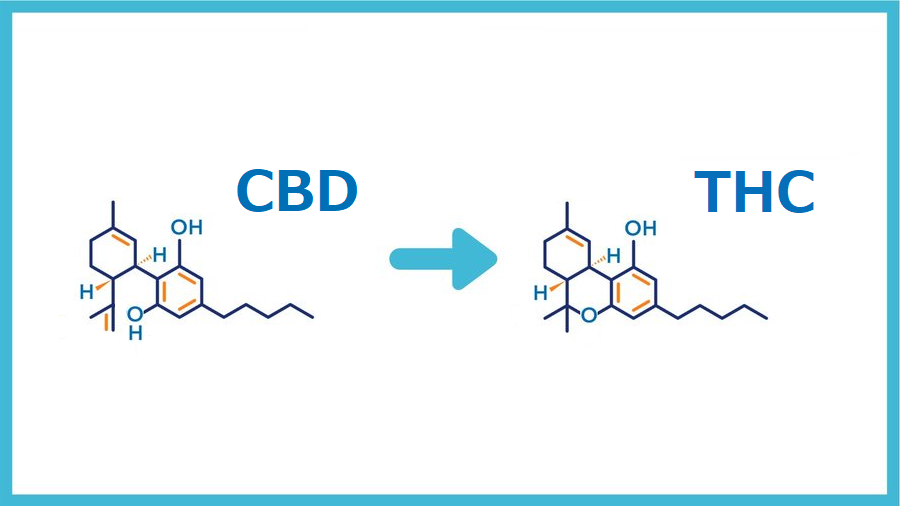
J. Nat. Prod. 2020, 83, 2894−2901 より
概要
カンナビジオールの化学反応性は、2つの二重結合の1つにフェノール基を付加することによって駆動される分子内環化を受ける能力に基づいています。
この環化の主な生成物は、Δ9-THC(トランス-Δ-9-テトラヒドロカンナビノール)およびΔ8-THC(トランス-Δ-8-テトラヒドロカンナビノール)です。
これらの2つのカンナビノイドは異性体であり、最初のカンナビノイドは頻繁に研究されている向精神薬と医薬品です。
異性体Δ8-iso-THC(trans-Δ-8-iso-テトラヒドロカンナビノール)およびΔ4(8)-iso-THC(trans-Δ-4,8-iso-テトラヒドロカンナビノール)は、分子内環化の追加生成物として同定されています。
CBD(カンナビジオール)の反応性の可能な調節を調査するために、異なる溶媒でのルイス酸と非プロトン酸の使用が研究されてきました。
4つの異性体の完全なNMR分光学的特性が報告されています。高速液体クロマトグラフィー分析および反応混合物の1HNMRスペクトルを使用して、形成された化合物のパーセンテージ比を評価しました。
近年、フィトカンナビノイドへの関心が劇的に高まっています。 1940年に大麻から分離されたカンナビジオール(CBD)は、繊維用途の大麻種の中で最も豊富な植物性カンナビノイドの1つです。 CBDとΔ9-THC(トランス-Δ9-テトラヒドロカンナビノール)の構造的類似性にもかかわらず(図1)、CBDはカンナビノイド受容体に対するアゴニスト効果が低いです。特に、CB1およびCB2受容体(カンナビノイド受容体タイプ1および2)のアロステリックネガティブモジュレーターと見なされます。現在の証拠は、CBDが、アデノシン、グリシン、オピオイド、セロトニン、非エンドカンナビノイドGタンパク質共役型、ニコチン性アセチルコリン、および増殖因子活性化受容体などの特定の分子標的を介して薬理学的効果を発揮することを示しています。
さらに、CBDは抗けいれん薬、鎮痙薬、抗不安薬、抗うつ薬、抗リウマチ性関節炎、および神経保護作用を示します。最近、CBDがGPR3、GPR6、GPR12などのGタンパク質共役型オーファン受容体のインバースアゴニストであることが実証され、アルツハイマー病、パーキンソン病、癌、不妊症に対するCBDの新しい治療用途が示唆されています。Δ9-THC主要な精神活性効果を持つカンナビスサティバの重要な化合物です。薬理学的観点から、Δ9-THCはCB1、精神活性効果のモジュレーター、およびCB2、免疫学的および抗炎症効果のモジュレーター両方のカンナビノイド受容体の部分アゴニストです。 Δ9-THCの精神活性効果には、不安、妄想、知覚の変化、および認知障害が含まれます。これらすべてのCB1媒介効果は、GABA(γ-アミノ酪酸)/グルタミン酸作動性神経伝達およびドーパミン放出の摂動によって引き起こされ、とりわけ、それらは一般に急性、一過性、および自己制限的です。さらに、マウスモデルではΔ9-THCの急性毒性が低いことも観察されています。最後に、Δ9-THC投与後、運動低下、低体温、カタレプシー、鎮痛、および食物摂取の増加が報告されています。
THC骨格を作成するためにCBDの分子内環化を誘発する可能性はよく知られています。 CBD、Δ9-THC、およびその異性体の間の活性の点で顕著な違いがあるため、
(a)この反応の実現可能性、(b)その選択性、および(c)この変換を監視するための効率的で迅速な方法の可用性を調査することにしました。
したがって、CBDをルイス酸と非プロトン酸で処理し、得られた混合物の組成を高速液体クロマトグラフィー(HPLC)または直接NMRスペクトル分析を使用して評価しました。
結果と考察
文献によると、CBDの環化反応は、特定の二重結合の酸触媒による活性化に続いて起こるようです。 ジヒドロベンゾピラン環部分は、二重結合の1つを持つフェノール基の1つの内部エーテル形成によって形成されます。 CBD構造の2つの二重結合は、2つの異なる化合物の形成に関与します(スキーム1)。 Δ8二重結合で活性化が起こった場合、生成物はTHC足場を示します(Δ9-THC、パスb)。 そうでなければ、Δ1二重結合の活性化はiso-THC足場の形成につながります(Δ8-iso-THC、パスa)。
後者の環化ははるかに少ない頻度です。 ただし、酸性条件は、対応する熱力学的に安定した化合物、それぞれΔ8-THCおよびΔ4(8)-iso-THCへのさらなる異性化の原因となります。
Δ9-THCとその誘導体は広く探求され、主要な精神活性大麻成分として認識されていますが、iso-THC異性体はほとんど注目されていません。このため、特に完全なNMRデータの提供に関して、文献のギャップを埋めたいと考えています。 CBD環化の感受性と選択性を調査するために、さまざまな溶媒でのルイス酸と非プロトン性酸の使用、温度と反応時間の変化など、さまざまな反応を実行しました(スキーム2)。ルイス酸は、BF3・OEt2の記録された使用から始めて、最初に評価されました。
データは、CH2Cl2中でBF3・OEt2との反応を低温で行うと、主生成物としてΔ9-THCが得られることを示唆していますが、温度と反応時間を長くすると、より安定したΔ8-THCが優先的に形成されます。結果はこのアサーションをサポートします(表1、エントリー1および2)。温度を下げると、収量も低下します(表1、エントリー3)。他の溶媒を使用すると、さまざまな程度の選択性が得られました。特に、トルエンはCH2Cl2と同様の結果を示しましたが、iso-THCは常にΔ8-およびΔ9-THCを伴いました(表1、エントリー4および5)。 MeCNで-10°Cで6時間行った反応により、微量のΔ4(8)-iso-THCを伴う主生成物としてΔ8-iso-THCが生成されました(表1、エントリー7)。
プロセスの収率と選択性を高めるために、他のルイス酸が積極的に環化を誘発する可能性があるという仮説に従って、さまざまな酸を使用した一連のテストを実施しました。異性化なしでΔ9-THC形成の高収率を示したTMSI(トリフルオロシリルヨウ化物)の使用に関する肯定的な文献結果から始めて、TMSOTf(トリメチルシリルトリフラート)を酸性試薬としてテストしました。予想に反して、CH2Cl2またはトルエンの場合、Δ8-THCの形成に対して高い親和性を示しました。
低温でも溶媒として使用されました(表1、エントリー8および9)。
CH2Cl2中のIn(OTf)3は、BF3・OEt2よりも高い収率で低温でCBDをΔ9-THCに変換しました(表1、エントリー10)。以前のテストと同様に、温度が高いほど、熱力学的に安定した異性体がより高い収率で生成されました(表1、エントリー11)。トルエンを使用すると、選択性は優れた収率でΔ8-THCの形成にシフトしました(表1、エントリー13)。 CH2Cl2にZnBr2を使用しても、室温でも環化反応は促進されませんでしたが(表1、エントリー14)、TiCl4はBF3と同様の傾向を示しました(表1、エントリー15)。 AlCl3、AgOTf、およびTi(OiPr)4の活性も調査されましたが、注目すべき結果はありませんでした。これらの結果を考慮すると、環化を誘発するために使用されるルイス酸の特性に基づいて、考えられる異性体のいずれかの固有の優先的な形成経路を決定することはできません。続いて、非プロトン性酸スクリーニングを実施しました(表2)。 CBD変換の最良の結果は、HCl、pTSA(p-トルエンスルホン酸)、およびCSA(カンファースルホン酸)で得られました。報告されているように、CH2Cl2中の9 pTSAは、唯一の生成物としてΔ8-THCの形成を直接もたらしました(表2、エントリー2)。溶媒の性質は明らかに反応結果に影響を及ぼしました。 n-ヘキサン中での反応により、Δ9-THC、Δ8-THC、およびΔ8-iso-THCの混合物が1:5:1の比率で得られ(表2、エントリー3)、トルエン中での反応により高い選択性が得られました。 。 pTSAは、溶媒と反応時間に応じて、さまざまな異性体をさまざまな割合で生成しました(表2、エントリ2〜5)。 Δ9-THCおよびΔ8-THC形成の最良の選択性は、それぞれトルエンおよびCH2Cl2で得られました。逆に、トルエン中の触媒量10%ミリモルの酸を使用すると、反応時間が長くなり、結果が熱力学的異性体にシフトしたため、二重結合がほぼ完全に異性化されました(表2、エントリー6)。興味深いことに、CSAはCBDのΔ9-THCへの環化を促進し、反応時間に関係なく完全な選択性と満足のいく収率を示しました(表2、エントリー7)。他の非プロトン性酸は、CBD変換に対してより悪い結果をもたらしました(表2、エントリー8-15)。いくつかのΔ4(8)-iso-THC形成が3つのケースで検出され(表1、エントリー5および7;表2、エントリー8)、この化合物が分離され、特性評価されました。
実験結果は、トルエンがCBDをTHC異性体に変換するのに最も適した溶媒であることを示しています。この溶媒は、他の実験条件(反応温度と酸環化促進剤の性質)による異性体の選択性に特に影響を与えます。温度を上げると、二重結合の活性化の選択性が低下し、対応する最も安定した異性体の形成が促進されます。ルイス酸BF3・OEt2、In(OTf)3、およびTMSOTfは効果が証明されており、Δ8-THCと生成物の混合物の主要な形成に影響を与えました。非プロトン性酸に関しては、pTSAは反応を促進し、反応時間に応じてΔ9-およびΔ8-THCを選択的に生成します。 CSAは興味深い環化誘導物質として出現し、容易にアクセスできる反応条件を通じてΔ9-THCを良好な収率で与えます。これらの有望なスクリーニング結果により、CSAの影響がより徹底的に調査されました。したがって、溶媒、温度、および時間は変数と見なされました。トルエンを溶媒として室温で使用すると、未反応のCBDを伴うΔ9-THCの収率が61%になりました(表2、エントリー7)。より長い反応時間は、Δ9-THCの異性化と化合物の分解をもたらしました(表3、エントリー3)。温度を上げると、時間の経過とともにΔ8異性体が濃縮された混合物が形成されました(表3、エントリー4〜8)。温度を上げると、CBDの変換時間と異性化がさらに大幅に減少し、反応速度論的反応生成物としてΔ9-THCが得られました(表3、エントリー9および10)。 CH2Cl2では、反応はより速く、Δ9-THC形成に対して選択性が低くなりました(表3、エントリー11〜13)。 n-ヘキサンとMTBE(t-ブチルメチルエーテル)は、短い反応時間でもTHC異性体の形成に対して顕著な優先的選択性を示すことなく、高度なCBD変換を誘発しました(表3、エントリー14および15)。すべての実験で、MTBEで反応を行った場合を除いて、iso-THC異性体は検出されませんでした(表3、エントリー15)。
トルエンは最高の選択性を示しました。しかし、反応時間が長いのは欠点のようでした。 CH2Cl2はこの点で有望であるように見えましたが、異性化の蔓延を避けるために反応の継続的なモニタリングが必要でした。
Δ9-THC、Δ8-THC、Δ8-iso-THC、およびΔ4(8)-iso-THCは、NMRデータを使用して完全に特徴付けられました。
完全な1Hおよび13CNMR割り当て(表4-6)が決定されました
1Dおよび2DNMRスペクトル(1Hおよび13C NMR、相関分光法(COSY)、異核単一量子コヒーレンス(HSQC)、および異核多重結合相関(HMBC))に基づいています。
データは、文献で入手可能なものと比較されました。診断的で識別可能なNMRピークにより、粗反応混合物内の分子内環化に由来する化合物の識別が可能になり、また、積分比から組成パーセンテージを決定することができます(図2)。 CDCl3では、Δ9-THCは、6.34 ppm(H-10)、3.23 ppm(H-10a)、2.22〜2.16 ppm(H-8)、および4.88 ppm(OH)の信号の存在によって特徴付けられます。 Δ8-THCの1HNMRデータは、H-10の2つの信号(3.21および2.19-2.15 ppm)を示していますが、H-10aによる信号は2.71ppmに存在します。オレフィン(H-8)とヒドロキシプロトンはそれぞれ5.45と4.63ppmに現れます。 Δ8-iso-THCの場合、対応する特性信号は、4.98 ppm(H-9)のダブレットと、それぞれプロトンH-3およびH-4に一致する3.49および2.37ppmの2つの信号です。 Δ4(8)-iso-THCのスペクトルは、4.29 ppm(H-3)で特徴的な信号を示しています。文献データ17に基づいて、HPLC法を使用してCBDの環化を追跡しました。 HPLCを介して監視された反応は、1H NMRデータ分析の組成に匹敵する反応混合物の組成を提供した。分析は、ASCENTIS RP-C18カラム(5μm×4.6×150 mm)で実行されました。圧力は101barに設定し、温度は0.95 mL / minの一定流量で40°Cに維持しました。グラジエント溶出法を使用して、228.8nmでUVスペクトルを記録しました。移動相は、A(H2O中の0.1%v / v HCOOH)とB(MeCN中の0.1%v / v HCOOH)の混合物で構成されていました。グラジエント溶出プログラムを30分の持続時間に適合させて、CBDの場合はRRT 1.00、Δ9-THCの場合はRRT1.28を得ました。 30分後、カラムを7分で100%Bでパージしました。その後、システムをこれらの条件下で3分間洗浄し、初期状態に戻しました。保持時間はCBD、23.63分でした。 Δ4(8)-iso-THC、29.62分; Δ9-THC、29.92分; Δ8-THC、30.77分;およびΔ8-iso-THC、30.77分(図3)。このメソッドにより、THC異性体からCBDを優れた方法で分離できました。特に、Δ8-THCとΔ8-iso-THCからΔ9-THCを認識することができました。しかし、ピークはかなり近かった。欠点は、同様の保持時間を持つΔ9-THC/Δ4(8)-iso-THCとΔ8-THC/Δ8-iso-THCのピーク間でより良い分解能を得ることが依然として困難であったことでした。
このため、HPLCの結果は常に1HNMRデータから得られた結果と比較されました。結論として、すべてのTHC異性体は、1Hおよび13CNMR分光法によって完全に特徴付けられました。分析方法は、反応の過程を監視するために最適化されました。特に、室温(RT)で96時間のトルエン中のCSAおよびRTで48時間のトルエン中のpTSAが、Δ9-THCの選択的形成に最適な条件であることがわかりました。
CH2Cl2中のTMSOTf(-10°Cで6時間)、トルエン中のIn(OTf)3(0°Cで24時間)、CH2Cl2中のpTSA(RTで36時間)、およびトルエン中のCSA(40°Cで96時間)により、高収率でΔ8THCが選択的に得られました。 。
トルエン中でBF3・OEt2を使用すると、反応温度に応じて、iso-THC異性体が形成されました。 -10°Cでは、Δ9-THCとΔ8-iso-THCの分離可能な混合物が得られましたが、0°Cへの温度上昇により、結果は対応する最も安定した異性体であるΔ8-THCとΔ4(8)-isoTHCにシフトしました。 。 CBDは、天然のアルケンとフェノールの化学反応性に対処し、活用することを可能にする挑戦的な基質です。
実験セクション
一般的な実験手順。特に明記しない限り、試薬と溶媒はSigma-Aldrich(ミラノ、イタリア)、Fluorochem(ハドフィールド、英国)、またはTCI(Zwijndrecht、ベルギー)から購入し、さらに精製することなく使用しました。すべての反応は、オーブン乾燥したガラス器具と乾燥溶媒中で窒素雰囲気下で実施し、シリカゲル(Merckプレコート60F254プレート)でのTLC、UV光(254 nm)またはモリブデン酸セリウム染色(ハネシアン染色)でモニターしました。 。分析用HPLCは、ASCENTIS RP-C18カラム(5μm×4.6×150 mm)で実施しました。圧力は約101barに設定し、温度は0.95 mL / minの一定流量で40°Cに維持しました。グラジエント溶出法を使用して、228nmでUVスペクトルを記録しました。移動相は、A(H2O中の0.1%v / v HCOOH)とB(MeOH中の0.1%v / v HCOOH)の混合物で構成されていました。勾配は、30分で60%Bから90%Bまで直線的にプログラムされました。フラッシュカラムクロマトグラフィー(FCC)は、固定相としてシリカゲル(240〜400メッシュ、Merck)を使用して実施しました。 1HNMRスペクトルはBrukerAvance Spectrometer300または400MHzで記録され、化学シフトは残留CDCl3、メタノール-d4、またはアセトン-d6と比較して報告されています。 13C NMRスペクトルは同じ機器(101 MHz)で記録され、化学シフトは残留CDCl3、メタノール-d4、またはアセトン-d6と比較して報告されています。すべての1Dおよび2DNMRスペクトルは、Bruker Topspin1.3で利用可能な標準パルスシーケンスを使用して取得されました。プロトンおよび炭素共鳴の化学シフト(δ)は、内部標準として使用されるTMSに対する100万分の1(ppm)で示されます。 1H NMRのデータは、化学シフト(δ/ ppm)、多重度、および結合定数(Hz)として報告されます。多重度は次のように報告されます:s =一重項、d =二重項、t =三重項、m =多重項、およびbr s =広い一重項。 13C NMRのデータは、化学シフト(δ/ ppm)で報告されています。 MSスペクトルは、Waters Micromass四重極飛行時間型マイクロ質量分析計でエレクトロスプレーイオン化(ESI)技術を使用して記録され、HR-ESI質量スペクトルはFT-ICR APEXII機器(Bruker Daltonics)で記録されました。 EI質量スペクトルは、VG 70-70EQで6kEvのイオン化電圧で記録されました。旋光度の値は、Jasco P-1030旋光計で、ナトリウムD線の波長λ= 589nmを使用して20°Cで測定しました。
ルイス酸または非プロトン性酸を使用する一般的な手順。すべての反応は、異なる無水溶媒中、異なる温度で窒素雰囲気下で実施された。指定された温度のCBD撹拌溶液(詳細は以下を参照)に、対応するルイス酸または非プロトン酸をゆっくりと加え、混合物を撹拌した。反応を飽和NaHCO 3水溶液でクエンチし、30分間撹拌し、飽和NaHCO 3水溶液およびブラインで洗浄した。有機相をNa2SO 4で乾燥し、濾過し、減圧下で蒸発させた。すべての反応は、モリブデン酸セリウム染色によって開発されたTLC(CH2Cl2 / n-ヘキサン1:3)によってモニターされ、粗生成物は、CDCl3およびHPLCでの1HNMR分光法によって分析されて組成が決定されました。すべての残留物をシリカゲル上のFCC(CH2Cl2 / n-ヘキサン1:3)で精製し、4つの可能なTHC異性体を提供しました。
CBD。 [α] D20 −113(c 1、EtOH); [HPLC ASCENTIS C18; RT CBD = 23.63分]; 1Hおよび13CNMRデータは表4を参照してください。 HRMS(ESI)m / z [M + Na] + 337.2137(C21H30O2Naの計算値、337.213)。
Δ9-THC。 [α] D20 −159(c 1、CHCl3); [HPLC ASCENTIS C18; RTΔ9-THC= 29.92分]; 1Hおよび13CNMRデータは表5を参照してください。 HRMS(ESI)m / z [M + Na] + 337.2132(C21H30O2Naの計算値、337.2138)。
Δ8-THC。 [α] D20 −238(c 1、CHCl3); [HPLC ASCENTIS C18; RTΔ8-THC= 30.77分]; 1Hおよび13CNMRデータは表5を参照してください。 HRMS(ESI)m / z [M + Na] + 337.2136(C21H30O2Naの計算値、337.2138)。
Δ8-iso-THC。 [α] D20 -249(c 1、CHCl3); [HPLC ASCENTIS C18; RTΔ8-iso-THC= 30.77分]; 1Hおよび13CNMRデータは表6を参照してください。 HRMS(ESI)m / z [M + Na] + 337.2141(C21H30O2Naの計算値、337.2138)。
Δ4(8)-iso-THC。 [α] D20 −236(c 1、CHCl3); [HPLC ASCENTIS C18; RTΔ4(8)-iso-THC = 29.62分]; 1Hおよび13CNMRデータは表6を参照してください; HRMS(ESI)m / z [M + Na] + 337.2133(C21H30O2Naの計算値、337.2138)。
BF3・OEt2触媒反応(表1)。ルイス酸の一般的な手順で指定されているように反応を行った。条件(表1、エントリー1):CBD(315 mg、1ミリモル);溶媒、無水CH2Cl2(5 mL); T = −10°C; BF3・OEt2(151μL、1.2ミリモル);反応時間、4時間。収量:Δ9-THC、138 mg(44%); Δ8-THC、4 mg(1%); Δ8-iso-THC、11 mg(3%)。条件(表1、エントリー2):CBD(315 mg、1ミリモル);溶媒、無水CH2Cl2(5 mL); T = 0°C; BF3・OEt2(151μL、1.2ミリモル);反応時間、6時間。収量:Δ9-THC、5 mg(2%); Δ8-THC、164 mg(52%)。条件(表1、エントリー3):CBD(315 mg、1ミリモル);溶媒、無水CH2Cl2(5 mL); T = −78〜−30°C; BF3・OEt2(151μL、1.2ミリモル);反応時間、48時間。収量:Δ9-THC、32 mg(10%); Δ8-THC、35 mg(11%); Δ8-iso-THC、16 mg(5%)。条件(表1、エントリー4):CBD(156 mg、0.5ミリモル);溶媒、無水トルエン(2.5 mL); T = −10°C; BF3・OEt2(76μL、0.6ミリモル);反応時間、3時間。収量:Δ9-THC、64 mg(41%); Δ8-THC、3 mg(2%); Δ8-iso-THC、45 mg(29%)。条件(表1、エントリー5):CBD(316 mg、1ミリモル);溶媒、無水トルエン(5 mL); T = 0°C; BF3・OEt2(151μL、1.2ミリモル);反応時間、6時間。収量:Δ8-THC、115 mg(36%); Δ4(8)-iso-THC、83 mg(26%)。条件(表1、エントリー7):CBD(315 mg、1ミリモル);溶媒、無水MeCN(5 mL); T = −10°C; BF3・OEt2(151μL、1.2ミリモル);反応時間、6時間。収量:Δ8-THC、16 mg(5%); Δ8-isoTHC、95 mg(30%); Δ4(8)-iso-THC、17 mg(5%)。
TMSOTf触媒反応(表1)。ルイス酸の一般的な手順で指定されているように反応を行った。条件(表1、エントリー8):CBD(315 mg、1ミリモル);溶媒、無水CH2Cl2(5 mL); T = −10°C; TMSOTf(217μL、1.2ミリモル);反応時間、6時間。収量:Δ8-THC、293mg(93%)。条件(表1、エントリー9):CBD(80 mg、0.25ミリモル);溶媒、無水トルエン(1.25 mL); T = −10°C; TMSOTf(91μL、0.5ミリモル);反応時間、6時間。収量:Δ9-THC、10 mg(12%); Δ8-THC、61 mg(75%)。 In(OTf)3触媒反応(表1)。ルイス酸の一般的な手順で指定されているように反応を行った。条件(表1、エントリー10):CBD(317 mg、1ミリモル);溶媒、無水CH2Cl2(5 mL); T = −10°C; In(OTf)3(675 mg、1.2ミリモル);反応時間、6時間。収量:Δ9THC、165 mg(52%); Δ8-THC、18 mg(6%); Δ8-iso-THC、12 mg(4%)。条件(表1、エントリー11):CBD(317 mg、1ミリモル);溶媒、無水CH2Cl2(5 mL); T = 0°CからRT; In(OTf)3(58 mg、0.1ミリモル);反応時間、48時間。収量:Δ8-THC、228mg(72%)。条件(表1、エントリー13):CBD(156 mg、0.5ミリモル);溶媒、無水トルエン(2.5 mL); T = 0°C; In(OTf)3(563 mg、1ミリモル);反応時間、24時間。収量:Δ8-THC、153mg(98%)。
TiCl4触媒反応(表1)。反応は、ルイス酸の一般的な手順で指定されているように実行されました。条件(表1、エントリー15):CBD(315 mg、1ミリモル);溶媒、無水CH2Cl2(5 mL); T = −10°C; TiCl4(167μL、1.2ミリモル);反応時間、6時間。収量:CBD、38 mg(12%); Δ9-THC、108 mg(34%); Δ8-THC、27 mg(9%)。
HCl触媒反応(表2)。非プロトン性酸の一般的な手順で指定されているように反応を行った。
条件(表2、エントリー1):CBD(156 mg、0.5ミリモル);溶媒、H2O(1.6 mL); T = RT; HCl 37%(1.6 mL);反応時間、72時間。収量:Δ8-THC、89mg(57%)。
pTSA・H2O触媒反応(表2)。非プロトン性酸の一般的な手順で指定されているように反応を行った。
条件(表2、エントリー2):CBD(154 mg、0.5ミリモル);溶媒、無水CH2Cl2(2.5 mL); T = RT; pTSA・H2O(189 mg、1ミリモル);反応時間、36時間。収量:Δ8-THC、145mg(94%)。条件(表2、エントリー3):CBD(155 mg、0.5ミリモル);溶媒、n-ヘキサン(2.5 mL); T = RT; pTSA・H2O(190 mg、1ミリモル);反応時間、36時間。収量:Δ9-THC、20 mg(13%); Δ8-THC、102 mg(66%); Δ8-iso-THC、20 mg(13%)。条件(表2、エントリー5):CBD(318 mg、1ミリモル);溶媒、無水トルエン(5 mL); T = RT; pTSA・H2O(386 mg、2ミリモル);反応時間、48時間。収量:Δ9-THC、262 mg(82%); Δ8-THC、34 mg
(11%)。条件(表2、エントリー6):CBD(79 mg、0.25ミリモル);溶媒、無水トルエン(1.25 mL); T = RT; pTSA・H2O(6 mg、0.025ミリモル);反応時間、96時間。収量:Δ9-THC、7 mg(9%); Δ8-THC、70 mg(89%)。
CSA触媒反応(表2)。反応は、非プロトン性酸の一般的な手順で指定されているように実行されました。
条件(表2、エントリー7):CBD(79 mg、0.25ミリモル);溶媒、無水トルエン(1.25 mL); T = RT; CSA(117 mg、0.5ミリモル);反応時間、96時間。収量:CBD、28 mg(36%); Δ9-THC、48 mg(61%)。
H2SO4触媒反応(表2)。非プロトン性酸の一般的な手順で指定されているように反応を行った。
条件(表2、エントリー8):CBD(315 mg、1ミリモル);溶媒、無水CH2Cl2(5 mL); T = 0°C; H2SO4、98%(54μL、1ミリモル);反応時間、72時間。収量:Δ8-THC、16 mg(5%); Δ8-iso-THC、13 mg(4%); Δ4(8)-iso-THC、37 mg(11%)。
CSAスクリーニング反応の一般的な手順(表3)。
手順はルイス酸の一般的な手順で説明した手順と同じですが、反応をEtOAcで希釈してクエンチし、モリブデン酸セリウム染色で生成したTLC(n-ヘキサン/ EtOAc7:3、2回溶出)でモニターしました。粗生成物を、CDCl3およびHPLCでの1H NMR分光法によって分析して、組成を決定した。条件:CBD(1当量); CSA(2当量);溶媒、トルエン(0.2 M)または表3に指定されているとおり。Tは表3に指定されているとおり。
コメント